| Up Contents |
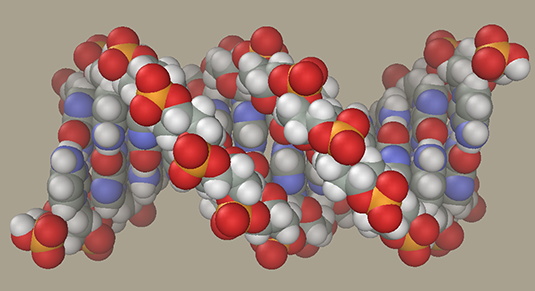
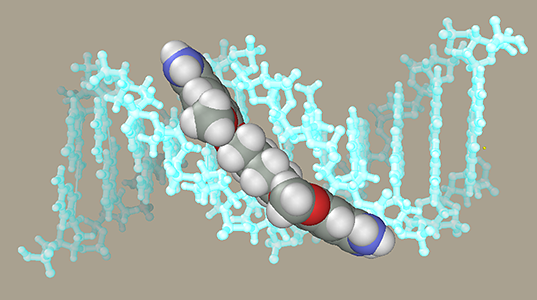
DNA-ligand modelingLet us generate a DNA model with a ligand in the minor groove. DNA
Open the Chain Editor
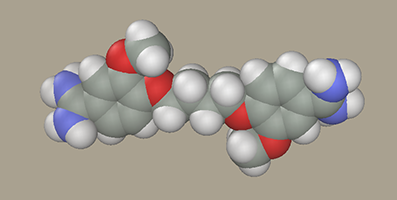
LigandSelect the Chain Editor B-DNA ligands section. Several residue samples are available there to construct ligands of the pentamidine series (J Med Chem, 1992, v. 35, pp.431-438) as well as residues to construct the alkyl bridge. For example, select AmPh(OMe)OCH2- L and click -CH2- twice and AmPh(OMe)OCH2- R once. Note that two -CH2- were selected, while the resulting model has four methylene groups. The point is that the methylene groups attached to oxygen have partial atomic charges different from those in carbohydrates. Accordingly, such methylene groups were added to the model of the ligand terminal residue (AmPh(OMe)OCH2-) in advance.
Rotate the ligand OMe groups so that they face the same direction.
This can be done by a short molecular dynamics simulation with restraints
imposed on the model. The molecule should remain extended and the OMe
groups should face the same direction. Invoke the Geometry Editor
Invoke the Molecular Dynamics panel
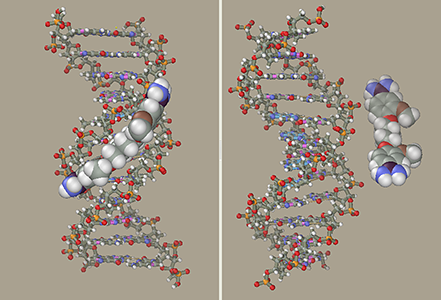
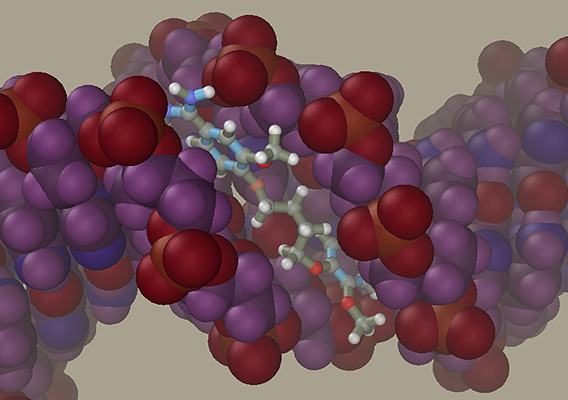
DNA-Ligand ComplexNow add the DNA model generated previously to the ligand model. Select the Add command in the File menu. At this moment, the initial and added models can be moved relative to each other, but we will not do it and press the OK button. Our ligand model has several selected atoms (if not, at least one of them should be selected with the right mouse). The molecules containing at least one selected atom can be moved relative to the rest of the model using a mouse with both buttons depressed. Place the ligand in front of the central part of DNA to face the minor groove using the Ctrl and Shift buttons and mouse. Avoid introducing the ligand directly into the minor groove, since this will cause strong collisions between atoms. Instead, place the ligand at some distance and use restraints to get it in desired place. Imagine that you attach rubber bands to the ligand and DNA, after which they contract and drive the ligand into the groove.
DNA is a relatively flexible construction. It can be deformed if the ligand runs into it at high speed. To avoid it, freeze DNA to allow movement of ligand atoms only.
Using the Geometry panel
The calculation can be accelerated by minimizing the window with the model, since showing the movement takes much processor time. After the preliminary optimization, the simulation can be started with no restrains or frozen molecules. The molecular dynamics in vacuum is practicable at low temperatures only, since normal temperature can destroy such crude models. Such molecular dynamics is an optimization approach to generate and roughly evaluate a model. Models with explicit solvents are recommended for simulations under normal conditions.
|
| Up Contents |
| Copyright © 2006-2010 Agile Molecule. All rights reserved |