| Up Contents |
Small Molecule ModelDraw molecule with a mouse
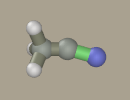
As an example, let us generate a model of acetonitrile molecule.
Open the Free drawing panel in the Build > Free
drawing menu or by pressing the toolbar button
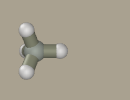
Select methane as a primer for the model by clicking the
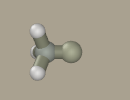
Mouse drawing from scratch is possible; however, using kernel is beneficial since it sets the scale. Holding Shift+Ctrl left-click the hydrogen atom to transform it to carbon.
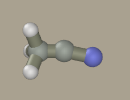
Select nitrogen in the panel and drag it from the carbon atom with the left mouse button (while holding Shift+Ctrl).
Select the triple bond and click the C-N bond. The bond has become green, which denotes triple bonds in the program. The bond order should not be necessarily specified. It is not included in quantum mechanical calculations; while in molecular mechanical calculations, the bond type is usually determined by the types of atoms that form the bond. Nevertheless, it is recommended to specify the bond order. First, it is marked by color on the screen and, second, the bond order determines how many hydrogen atoms should be added after pressing the Add hydrogen button. In this example, there is no need to press this button, since all atoms are in place.
Geometry optimization
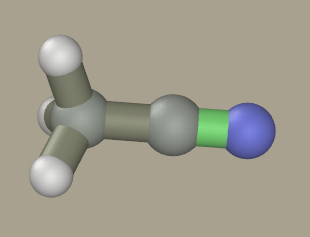
Since the model was hand-drawn, it is untidy. Its conformation can be
optimized. Rapid preliminary optimization can be done by semiempirical
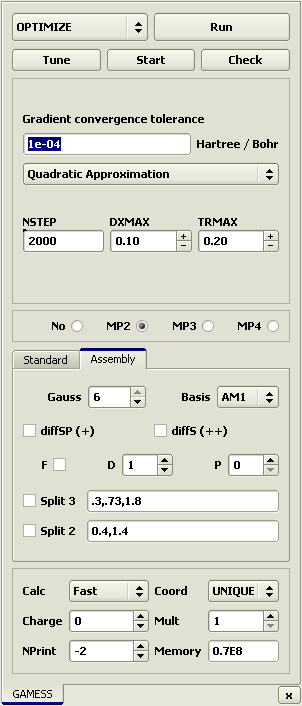
quantum chemical methods. Open the GAMESS panel in the
Tools > PC GAMESS or by pressing the toolbar button
It is recommended to press the Check button prior to quantum mechanical calculation to test the validity of the input data. This dummy calculation obviates the most common input errors. WARNING. The GAMESS program is not included in the Ascalaph package. According to the manufacturer's license, it cannot be included in other software packages and can be downloaded from the Laboratory of Chemical Cybernetics at Moscow State University. In order to install it in Ascalaph, unpack the downloaded file and transfer pcgamess.exe and DLLs to the Ascalaph/GAMESS folder. The optimization is started by the Run button. After several seconds, the molecule becomes symmetrical.
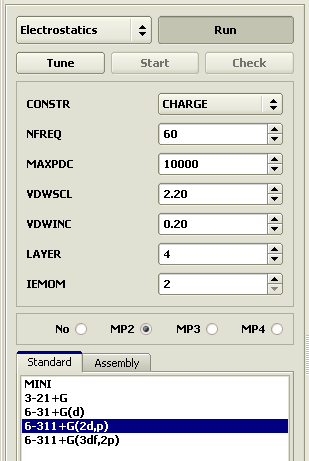
Potential Derived ChargesIf a model should be used in molecular mechanics calculations, partial electric charges should be specified for the atoms. For accurate calculations, use basis of at least the 2d quality with an account of electron correlation. Select the Electrostatics calculation. In the Standard panel, set the 6-311+G(2d,p) basis and MP2 electron correlation. Do not forget to Check the data and then start the calculation.
Clicking atoms with the left mouse button shows their charge in the status bar. The final action required to generate a simple molecular mechanics model is to save it to a file and define the molecular mechanics type for each atom. Unfortunately, the type assignment has to be done manually. Drawing a model gives the program no information but the atomic element and bond order. That is why, the free drawing constructor should be used only when convenient molecules are not available or cannot be assembled from the available residues.
|
| Up Contents |
| Copyright © 2006-2008 Agile Molecule. All rights reserved |