| Next Up Previous Contents |
Chain editorChain editor is the main tool to generate molecular models. It allows chain molecules to be constructed from predefined residues. Clearly, this construction mode is not universal. A required residue can be missing in the database or the constructed molecule can be not polymeric. Nevertheless, we believe that it is the principal construction mode due to two reasons. First, it corresponds to the requirements of scientists studying biological molecules. Proteins and nucleic acids are large chain structures. Due to their size, regular structure, and scientific importance, they require special construction tools. Second, our constructor is oriented towards developing molecular mechanics models. Hence, we need a tool to associate each atom with its molecular mechanics type as well as with the partial charge corresponding to the selected force field. Such association is a nontrivial task for large models. Some systems make the association by automatically defining the type of each atom according to its environment. From the user's viewpoint, it is a very convenient way. Unfortunately, correct working of such programs can hardly be guaranteed. The rules for type definition from the environmental atoms are complex and many such rules are required. Type definition error is hard to identify in a large model. We believe that explicit definition of types and charges for each force field provides for the most reliable associations. Such association presets are stored in special files (Repository folder). Unfortunately, this technique also cannot guarantee the correct working considering a great data volume to be entered; still, it is more reliable.
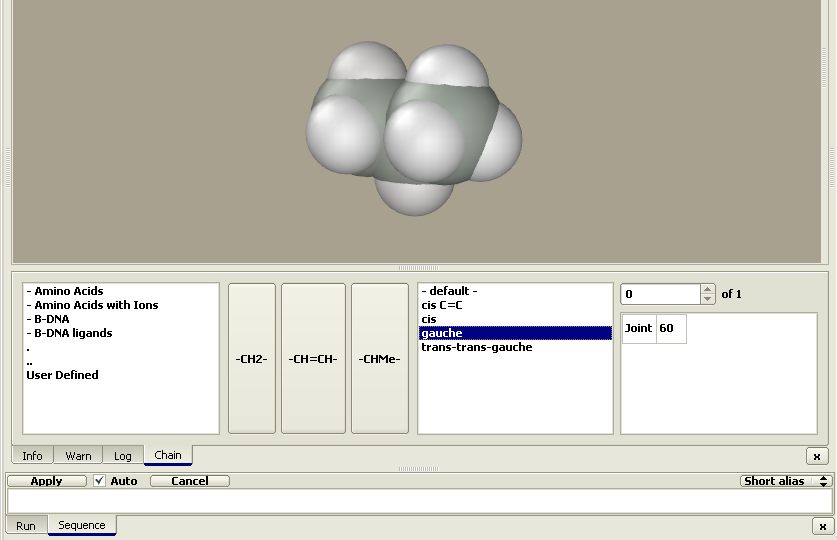
Chain editor can be invoked from the Build > Chain
menu or by pressing the toolbar button
The figure shows the Chain and Sequence panels. The buttons with residues hold the central position. If the Auto option is unchecked, pressing a residue button does not add the residue to the model. Instead, its name is added to the sequence text string, which is converted to a model after pressing the Apply button. In addition to pressing the residue buttons, the sequence string can be entered using keyboard or copied from a different program. Two alternative residue codes can be used, e.g., one-letter or three-letter codes for amino acids. Codes can be selected using the Alias button to the right of the sequence panel. The residue class can be selected in the window to the left of the residue buttons. Clicking on the desired class brings a set of buttons with the corresponding residues. The classes are arranged in a hierarchy of folders just as files on a PC. Double-clicking a folder name opens it, while double-clicking the ".." sign brings the user to the upper-level folder. Actually, these are subfolders in the Repository folder. Editing it allows the user to create individual sets of residues and conformational prototypes of chain molecules as described in the description of the Edit > Sequence menu. The Conformational type window can be found to the right of the residue buttons. The conformational type is defined by a set of parameters including the torsion angles (shown in the rightmost window). These parameters can be applied to each added residue or there may be sets of parameters applied to a sequence of added residues. For instance, the 'beta turn II' conformational type includes individual sets of parameters applied to each first, second, third, and fourth residue. The current step parameters are shown in the rightmost window. Let us construct several chain molecules. Example 1. Trans-gauche-trans hexanePress the –CH2– button. A propane molecule appears rather than methylene. The constructor is designed not to generate molecules with dangling bonds; instead, it generates a molecule composed of the desired residue with stub residues. The methyl group is commonly used as a stub residue. Accordingly, the methylene residue is presented in the Chain editor as propane: methylene with two methyl stubs (Me–CH2–Me). Pressing the –CH2– button once more yields a butane, i.e., the molecule was extended by a methylene group as follows:
Since no conformation type was chosen, this yielded a trans-butane (with a torsion angle of 180°). Let us append another methylene group in a gauche conformation. Select gauche as the Conformational type and press the –CH2– button once more. Select '– default –' as the Conformational type and press the –CH2– button to add another methylene group in a trans conformation. This yields a hexane molecule in a trans-gauche-trans conformation.
Comment on Force Field AssociationThe introduction of stub atoms in residues seeing as methyl groups and using them in assembly of chain molecules following the Me–CH2–Me + Me–CH2–Me mechanism is significant not only to avoid molecules with dangling bonds. The problem of molecular mechanics types associated with atoms is more important. A molecule assembled is already associated with a certain force field. The associated force field can be checked in the information window (menu Analyse > Information ). It looks like butane can be assembled from four methane molecules (CH4 + CH4 + CH4 + CH4) or from two ethane molecules (C2H6 + C2H6). However, what will be the atom type definition procedure in this case considering that methane and ethane have unique parameterization in some force fields? Butane molecule assembled from methane fragments would inherit wrong atom types. This problem is considered in detail in the Force Field Association section.
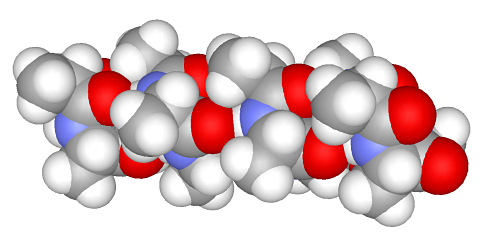
Example 2. Alpha-helixLet us assemble a biomolecule fragment with a desired secondary structure. Protein α-helix is the simplest example. Let us assemble it from alanine residues:
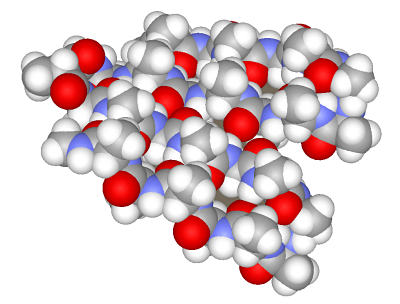
Example 3. Beta-sheetLet us now assemble a more complex structure of a β-sheet. It includes extended regions folded into a sheet due to type II β-turns. We will assemble a β-sheet from fragments including four amino acids for both the strand and turns.
|
| Next Up Previous Contents |
| Copyright © 2006-2009 Agile Molecule. All rights reserved |