| Next Up Previous Contents |
Molecular drawing
The Free drawing constructor of Ascalaph
can be accessed via the Build >
Free drawing menu or toolbar button
Edit mode works even if Free drawing panel is not active (overlapped by other panel). If Free drawing panel is closed then edit mode turns off. Hold Shift+Ctrl key combination during editing operations. Clicking on empty space creates a new atom. Clicking on an atom changes the element. Bonds can be dragged from atoms. If the button is released near a different atom, the bond is connected to it. Clicking on the bond sets its order. It is recommended to start molecular drawing from a Kernel formula rather than from scratch. Doing so sets of the model scale, which can be beneficial for its optimization later. In the standard mode, the current charge and elements correspond to those specified on the panel. In the Clone mode, after dragging a bond from an atom, the new atom has the same charge, element, and molecular mechanics type. This mode is convenient to draw a molecule composed of numerous atoms of the same type. Let us draw perfluoroalkane as an example. Assume we want CT type carbon atoms, F type fluorine atoms, and -0.1 charge. The atom model is to be saved in hin format.
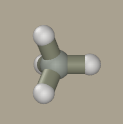
Select methane as a primer by clicking the
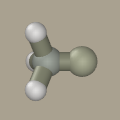
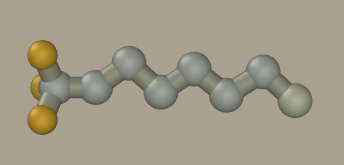
Click on hydrogen (while holding Shift+Ctrl) to convert it to carbon.
Save the model as C8F18.hin. Specify the desired elements, types, and charges: and reopen the file.
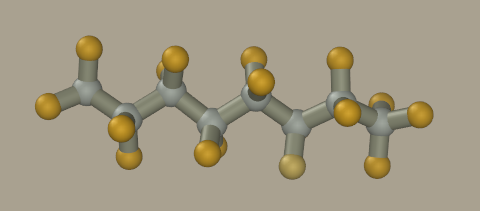
Set the F element and -0.1 charge on the panel. Activate the Clone mode and drag a chain of desired length.
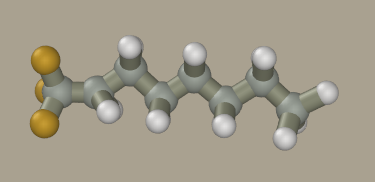
Deactivate the Clone mode and press the Add hydrogen button.
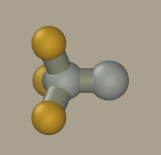
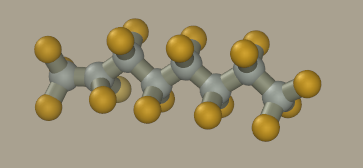
Now click on all hydrogen atoms to convert them to fluorine. We could mouse drag fluorine atoms from carbon atoms; however, the resulting geometry will be inaccurate and hard to optimize.
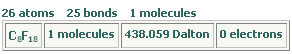
Set to the carbon element and +0.30 charge on the panel and click on the terminal carbon atom to correct its charge. The resulting model has -0.1 charge on the fluorine atom, +0.2 on the -CF2 carbon, and +0.3 on -CF3 terminal carbon atoms. Request the model information to make sure it was correctly made (Analyse > Information).
The model has correct molecular formula and zero total charge.
The atom model can be optimized to provide for a sensible geometry.
Preliminary optimization can be carried out in a crude force field.
The crude force field that requires no molecular mechanics type assignment
to atoms mode can be switched on by checking the Ad hoc option in the
Compute menu. Open the optimization panel in the
Computе > Optimize menu and press the Run button
Open the Quantum mechanics panel (toolbar button
Save the model. Since the fluorine atoms were not cloned, their molecular mechanics type was not specified, which should be done by editing the file. Other types and partial charges on atoms were specified correctly. This example demonstrates that the Free drawing constructor allows rapid drawing of molecules. However, it is not completely automatic and care should be taken to correctly specify the types and charges on atoms as well as bond orders. |
| Next Up Previous Contents |
| Copyright © 2006-2010 Agile Molecule. All rights reserved |